

By Erika Koutroumpa,
The Food and Drug Administration (FDA) qualifies a rare disease as one which affects fewer than 200,000 people, or as a condition that affects greater than 200,000 persons in the United States, but for which there is no reasonable expectation that pharmaceutical companies will recover the costs incurred during drug development following pharmaceutical sales. Examples include cystic fibrosis, and Huntington’s chorea; between 6,000 and 8,000 diseases in the United States meet this condition, affecting over 25 million patients in total.
The orphan drug act came into force in 1983, encouraging pharmaceutical companies to partake in research for therapies for rare diseases. Efforts began back in 1979, calling for a task force to deal with the growing “orphan drug problem”, namely the lack of medication for diseases with a small, affected population. An amendment to the Federal Food, Drug, and Cosmetic Act, due to the increase in thalidomide-related congenital diseases from the 1950s and led to the need for the orphan drug act to take place. Costs for drug development increased for all pharmaceutical companies, shifting the focus away from diseases with small affected populations and more towards widely-spread ailments with higher chances of profit. The initial form of the act gave participants multiple benefits including a waiver of approval fees, tax credits of up to 50 percent for research and development expenses, and a 7-year market exclusivity period, which led to more companies investing in orphan drug research.
So, what has been the impact of the Orphan Drug Act since its implementation 40 years ago? Until 2002, the number of orphan designations by the FDA grew to almost 1100 and approvals to 232, a number that provided treatment to an estimated 11 million patients. Currently, over 650 drugs have been approved, giving patients of rare diseases access to a greater variety of treatments. Even though not all orphan diseases have a cure yet, research in the field has greatly improved compared to nearly half a century ago and there is still great hope for the future. The enactment of the ODA inspired similar changes internationally in regions such as Europe, Australia, Singapore, and Japan.
Despite the significant boost in research on orphan drugs, the legislation has faced criticism. According to some skeptics, pharmaceutical companies are trying to take advantage of the benefits, by conducting more research on orphan drugs for diseases that bring the most profit. At least 95 of the approved orphan drugs were for cancer treatment; orphan drugs used to treat rare cancer are the most profitable. Additionally, despite monetary incentives intended to even out the lack of profit, due to the niche market of the drug, establishing a price that maximizes its profit is not prohibited by the ODA. High starting prices can be determined by the medication, making it difficult to access patients in poor economic conditions.
In 2010, orphan drugs represented 22% of total pharmaceutical sales and demonstrated an increased return on investment compared to non-orphan drugs. This type of medication has come to represent a significant source of global sales revenues thanks to ODA perks such as shorter clinical trial times, smaller clinical trial sizes, higher rates of regulatory success, and government financial incentives. The 2016 fee for an application, that includes clinical data is $2,374,200 and establishment fees are an additional $585,200. If the company gains Financial assistance from the Office of Orphan Products Development (OOPD), it is provided up to $250,000 per year for up to 3 years for phase 1 trials and up to $500,000 per year for up to 4 years for phase 2 and 3 trials. Hence, we have a government investment of 3-5 million dollars for a limited number of patients, and a minimum of 1 year of development.
In 2017, Kaiser Health News discovered, that drugmakers were manipulating the program to maximize revenues and protect niche markets for drugs used by millions. In several situations, FDA evaluators failed to demonstrate that they had determined how many patients may be treated by medicine being considered for orphan drug status. In over 60% of the cases, FDA reviewers failed to capture regulatory history information, including “adverse actions” from other regulatory bodies. The FDA’s evaluators are meant to follow two particular criteria — what percentage of patients are meant to be treated and if there is proof that the drug may mitigate their medical condition — and several orphan drug applications were flawed during their approval.
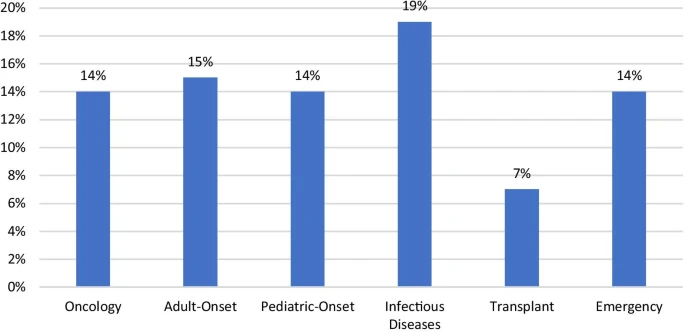
Furthermore, popular mass-market medications, such as the best-selling treatment in the world for rheumatoid arthritis, Humira, all received orphan designation despite already being utilized to treat common illnesses. In 2021, an amendment was introduced that requires corporations to demonstrate proof of financial damage caused by the production of orphan medicine in order to be given exclusivity and financial compensation. However, the results of this change are yet to be seen and this only reflects the new intention of the US government to revamp the orphan drug act.
In conclusion, Orphan Drug Act has brought a great change in how pharmaceutical companies in the US operate, encouraging research on diseases that would not have gotten any attention otherwise. Perhaps, though it has been too effective, due to frequent complaints about the morality of reduced clinical trials and the abuse of the perks by the pharmaceutical companies, making more profit than, with standard disease medication.
References
-
Government Investigation Finds Flaws In the FDA’s Orphan Drug Program. khn.org. Available here
-
Policymaking for Orphan Drugs and Its Challenges. journalofethics.ama-assn.org. Available here
-
Orphan drug. sciencedirect.com. Available here

