

Της Κωνσταντίνας Αργουδέλη,
Ως μεταβολισμό ορίζουμε το σύνολο των βιοχημικών αντιδράσεων που λαμβάνουν χώρα στο σώμα μας. Αυτός διαιρείται σε καταβολισμό και αναβολισμό. Με τον καταβολισμό διασπώνται τα συστατικά της τροφής στους δομικούς τους λίθους, δηλαδή σε αμινοξέα, σάκχαρα και λιπαρά οξέα, και παράγεται ενέργεια χρήσιμη για το κύτταρο. Με τον αναβολισμό γίνεται σύνθεση των μεγαλύτερων μορίων (μακρομορίων) του οργανισμού, όπως οι πρωτεΐνες, οι πολυσακχαρίτες, τα λιπίδια και τα νουκλεϊκά οξέα, με αξιοποίηση ενέργειας.
Οι αντιδράσεις αυτές καταλύονται από ένζυμα και υπόκεινται σε ρύθμιση, για να παράγουν ό,τι χρειάζεται ο οργανισμός κάθε στιγμή. Αν τα ένζυμα υπερλειτουργούν, υπολειτουργούν, ή απουσιάζουν πλήρως, προκύπτουν μεταβολικά νοσήματα. Τα μεταβολικά νοσήματα διακρίνονται σε μη κληρονομικά και κληρονομικά. Αναφορά θα γίνει στα κληρονομικά μεταβολικά νοσήματα, καθώς έχουν ξεκάθαρο γενετικό υπόβαθρο, σε αντίθεση με τα μη κληρονομικά, που εξαρτώνται από πολλούς παράγοντες.
Ένα σπάνιο μεταβολικό νόσημα είναι η Φαινυλκετονουρία (PKU), η οποία κληρονομείται με αυτοσωμικό υπολειπόμενο τρόπο. Προκαλείται από έλλειψη του ενζύμου υδροξυλάση της φαινυλαλανίνης, το οποίο μετατρέπει τη φαινυλαλανίνη σε τυροσίνη. Η έλλειψη έχει ως αποτέλεσμα τη συσσώρευση φαινυλαλανίνης στο αίμα και τον εγκέφαλο, δημιουργώντας εγκεφαλικές βλάβες. Το σημαντικότερο επακόλουθο της ασθένειας είναι η διανοητική υστέρηση. Ιδιαίτερη σημασία έχει η έγκαιρη διάγνωση, με το Guthrie test, τις πρώτες ημέρες μετά τη γέννηση του νεογνού. Και αυτό, γιατί η διανοητική υστέρηση μπορεί να προληφθεί με την τήρηση ενός διαιτολογίου απαλλαγμένο από φαινυλαλανίνη. Τα άτομα με Φαινυλκετονουρία πρέπει επίσης να αποφεύγουν την ασπαρτάμη, μια γλυκαντική ουσία που στον οργανισμό μετατρέπεται σε φαινυλαλανίνη και περιέχεται σε πολλά αναψυκτικά και τσίχλες. Το Guthrie test περιλαμβάνεται στο Εθνικό Πρόγραμμα Προληπτικού Ελέγχου Νεογνών που θεσπίστηκε το 1974 και γίνεται δωρεάν σε όλα τα νεογνά. Στον προληπτικό έλεγχο των νεογνών, σήμερα, περιλαμβάνονται επιπλέον η μέτρηση δραστικότητας του ενζύμου G6PD και ο έλεγχος του συγγενούς Υποθυρεοειδισμού και της Γαλακτοζαιμίας.
Το νόσημα που χαρακτηρίζεται από ανεπάρκεια του ενζύμου αφυδρογονάση της 6-φωσφορικής γλυκόζης (G6PD), γνωστό και ως κυαμισμός, είναι φυλοσύνδετο. Φυσιολογικά, το ένζυμο G6PD, που συμμετέχει στην οδό των φωσφορικών πεντοζών, μετατρέπει την 6-φωσφορική γλυκόζη σε 6-φωσφογλυκονο-δ-λακτόνη και παράγεται NADPH. Το NADPH είναι αναγωγικό μόριο, το οποίο ανάγει τη γλουταθειόνη και προστατεύει το κύτταρο από οξειδωτικές ουσίες όπως το υπεροξείδιο του υδρογόνου. Όταν υπάρχει έλλειψη του ενζύμου, δεν παράγεται επαρκής ποσότητα NADPH, με αποτέλεσμα να οξειδώνονται βασικά συστατικά των ερυθροκυττάρων και να προκαλείται αιμολυτική αναιμία. Η αναιμία συνήθως εκδηλώνεται μετά τη χρήση συγκεκριμένων φαρμάκων, την κατανάλωση κουκιών ή κάποια λοίμωξη.
Ένα ακόμη μεταβολικό νόσημα που κληρονομείται με φυλοσύνδετο τρόπο είναι η νόσος Fabry. Η νόσος Fabry είναι λυσοσωματική αθροιστική νόσος (LSD). Οφείλεται σε μερική ή ολική ανεπάρκεια του ενζύμου α-γαλακτοσιδάση Α, το οποίο φυσιολογικά αποδομεί έναν τύπο λιπιδίων. Η έλλειψή του, λοιπόν, οδηγεί σε συσσώρευση αδιάσπαστων γλυκοσφιγγολιπιδίων στους ιστούς και στο πλάσμα των ασθενών. Η συσσώρευση αυτή εμποδίζει την ομαλή λειτουργία των βασικών μεταβολικών διαδικασιών, οδηγώντας τα κύτταρα σε απόπτωση και δημιουργώντας βλάβες σε ζωτικά όργανα. Συμπτώματα της νόσου αποτελούν το διάχυτο άλγος (62%), οι δερματικές αλλοιώσεις (31%), οι γαστρεντερικές διαταραχές (19%), η νεφρική προσβολή (17%), ενώ ταυτόχρονα προκύπτουν και προβλήματα στους οφθαλμούς (11%). Η νόσος Fabry αντιμετωπίζεται με θεραπεία ενζυμικής αποκατάστασης, η οποία περιλαμβάνει την ενδοφλέβια χορήγηση του ενζύμου που δεν επαρκεί, κάθε δύο εβδομάδες.
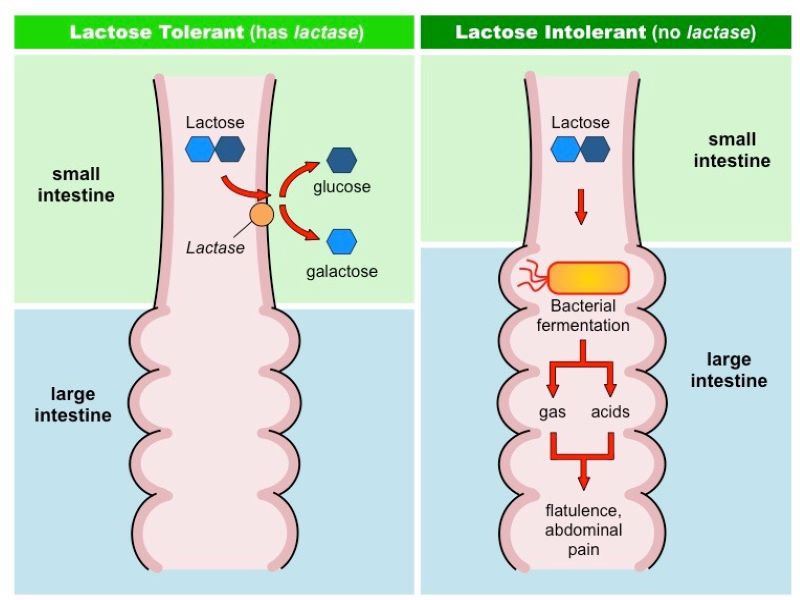
Τέλος, η δυσανεξία στη λακτόζη είναι ένα συχνό μεταβολικό νόσημα, με ποσοστό 30% στους Ευρωπαίους. Οφείλεται στην έλλειψη του ενζύμου λακτάση, το οποίο φυσιολογικά παράγεται από κύτταρα του λεπτού εντέρου και διασπά τον δισακχαρίτη λακτόζη στα δύο μονομερή του, τη γαλακτόζη και τη γλυκόζη. Επομένως, τα άτομα με ανεπάρκεια λακτάσης δεν μπορούν να μεταβολίσουν τη λακτόζη στο λεπτό έντερο, με αποτέλεσμα αυτή να μεταφέρεται αυτούσια στο παχύ έντερο, όπου τα βακτήρια τη μετατρέπουν σε λιπαρά οξέα βραχείας αλύσου. Η αντίδραση αυτή συνοδεύεται από παραγωγή αερίων, και συγκεκριμένα διοξειδίου του άνθρακα και μεθανίου. Τα συμπτώματα, λοιπόν, περιλαμβάνουν ναυτία, δυσπεψία, φούσκωμα στο στομάχι, κοιλιακό πόνο, καθώς και αποβολή αερίων. Για την αντιμετώπιση συστήνεται η αποφυγή της λακτόζης στη διατροφή, η οποία βρίσκεται σε μεγάλη ποσότητα στα γαλακτοκομικά.
Συνοψίζοντας, τα μεταβολικά νοσήματα προκαλούνται από πλήρεις ή μερικές ελλείψεις ενζύμων που συμμετέχουν στις μεταβολικές διεργασίες. Μερικά από αυτά αντιμετωπίζονται προληπτικά, ενώ άλλα με θεραπεία ενζυμικής αποκατάστασης. Σημείο-κλειδί αποτελεί η έγκαιρη διάγνωση με διενέργεια βιοχημικών εξετάσεων σε νεαρή ηλικία.
ΕΝΔΕΙΚΤΙΚΕΣ ΠΗΓΕΣ
- Phenylketonuria, www.nhs.uk. Διαθέσιμο εδώ
- Εργαστηριακές Εξετάσεις, ΕΠΠΕΝ. Διαθέσιμο εδώ
- Τι είναι η Ανεπάρκεια του ενζύμου G6PD… , ΕΠΠΕΝ. Διαθέσιμο εδώ
- Νόσος Fabry, Εργαστήριο Ιατρικής Γενετικής του Πανεπιστημίου Αθηνών. Διαθέσιμο εδώ
- Δυσανεξία στη λακτόζη;, Κουτκιά – Μυλωνάκη Πολυξένη, Βουτσά Διονυσία, www.hygeia.gr. Διαθέσιμο εδώ

